Generated from notebooks/python-api.ipynb
Kipoi python API
Quick start
There are three basic building blocks in kipoi:
- Source - provides Models and DataLoaders.
- Model - makes the prediction given the numpy arrays.
- Dataloader - loads the data from raw files and transforms them into a form that is directly consumable by the Model
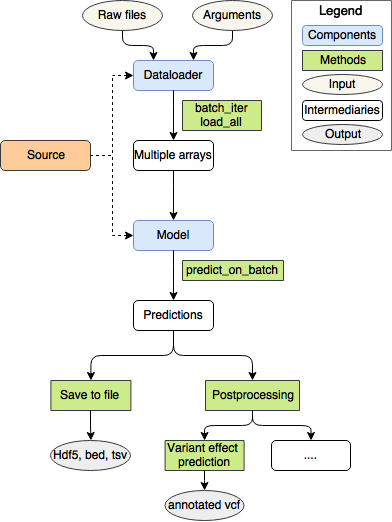
List of main commands
Get/list sources
- kipoi.list_sources()
- kipoi.get_source()
List models/dataloaders
- kipoi.list_models()
- kipoi.list_dataloaders()
Get model/dataloader
- kipoi.get_model()
- kipoi.get_dataloader_factory()
Load only model/dataloader description from the yaml file without loading the model
kipoi.get_model_descr()kipoi.get_dataloader_descr()
Install the dependencies
- kipoi.install_model_dependencies()
- kipoi.install_dataloader_dependencies()
import kipoi
Source
Available sources are specified in the config file located at: ~/.kipoi/config.yaml. Here is an example config file:
model_sources:
kipoi: # default
type: git-lfs # git repository with large file storage (git-lfs)
remote_url: [email protected]:kipoi/models.git # git remote
local_path: ~/.kipoi/models/ # local storage path
gl:
type: git-lfs # custom model
remote_url: https://i12g-gagneurweb.informatik.tu-muenchen.de/gitlab/gagneurlab/model-zoo.git
local_path: /s/project/model-zoo
There are three different model sources possible:
git-lfs- git repository with source files tracked normally by git and all the binary files like model weights (located infiles*directories) are tracked by git-lfs.- Requires
git-lfsto be installed. git- all the files including weights (not recommended)local- local directory containing models defined in subdirectories
For git-lfs source type, larger files tracked by git-lfs will be downloaded into the specified directory local_path only after the model has been requested (when invoking kipoi.get_model()).
Note
A particular model/dataloader is defined by its source (say kipoi or my_git_models) and the relative path of the desired model directory from the model source root (say rbp/).
A directory is considered a model if it contains a model.yaml file.
import kipoi
import warnings
warnings.filterwarnings('ignore')
import logging
logging.disable(1000)
kipoi.list_sources()
| source | type | location | local_size | n_models | n_dataloaders | |
|---|---|---|---|---|---|---|
| 0 | kipoi | git-lfs | /home/avsec/.kipoi/mo... | 1,2G | 780 | 780 |
s = kipoi.get_source("kipoi")
s
GitLFSSource(remote_url='[email protected]:kipoi/models.git', local_path='/home/avsec/.kipoi/models/')
kipoi.list_models().head()
| source | model | version | authors | contributors | doc | type | inputs | targets | postproc_score_variants | license | cite_as | trained_on | training_procedure | tags | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | kipoi | DeepSEAKeras | 0.1 | [Author(name='Jian Zh... | [Author(name='Lara Ur... | This CNN is based on ... | keras | seq | TFBS_DHS_probs | True | MIT | https://doi.org/10.10... | ENCODE and Roadmap Ep... | https://www.nature.co... | [Histone modification... |
| 1 | kipoi | extended_coda | 0.1 | [Author(name='Pang We... | [Author(name='Johnny ... | Single bp resolution ... | keras | [H3K27AC_subsampled] | [H3K27ac] | False | MIT | https://doi.org/10.10... | Described in https://... | Described in https://... | [Histone modification] |
| 2 | kipoi | DeepCpG_DNA/Hou2016_m... | 1.0.4 | [Author(name='Christo... | [Author(name='Roman K... | This is the extractio... | keras | [dna] | [cpg/mESC1, cpg/mESC2... | True | MIT | https://doi.org/10.11... | scBS-seq and scRRBS-s... | Described in https://... | [DNA methylation] |
| 3 | kipoi | DeepCpG_DNA/Smallwood... | 1.0.4 | [Author(name='Christo... | [Author(name='Roman K... | This is the extractio... | keras | [dna] | [cpg/BS24_1_2I, cpg/B... | True | MIT | https://doi.org/10.11... | scBS-seq and scRRBS-s... | Described in https://... | [DNA methylation] |
| 4 | kipoi | DeepCpG_DNA/Hou2016_H... | 1.0.4 | [Author(name='Christo... | [Author(name='Roman K... | This is the extractio... | keras | [dna] | [cpg/HepG21, cpg/HepG... | True | MIT | https://doi.org/10.11... | scBS-seq and scRRBS-s... | Described in https://... | [DNA methylation] |
Model
Let's choose to use the rbp_eclip/XRCC6 model from kipoi
MODEL = "rbp_eclip/XRCC6"
NOTE: If you are using python2, use a different model like MaxEntScan/3prime to following this example.
# Note. Install all the dependencies for that model:
# add --gpu flag to install gpu-compatible dependencies (e.g. installs tensorflow-gpu instead of tensorflow)
!kipoi env create {MODEL}
!source activate kipoi-{MODEL}
model = kipoi.get_model(MODEL)
Available fields:
Model
- type
- args
- info
- authors
- name
- version
- tags
- doc
- schema
- inputs
- targets
-
default_dataloader - loaded dataloader class
-
predict_on_batch()
- source
- source_dir
- pipeline
- predict()
- predict_example()
- predict_generator()
Dataloader
- type
- defined_as
- args
- info (same as for the model)
- output_schema
- inputs
- targets
-
metadata
-
source
- source_dir
- example_kwargs
- init_example()
- batch_iter()
- batch_train_iter()
- batch_predict_iter()
- load_all()
model
<kipoi.model.KerasModel at 0x7f95b27af2b0>
model.type
'keras'
Info
model.info
ModelInfo(authors=[Author(name='Ziga Avsec', github='avsecz', email=None)], doc='\'RBP binding model from Avsec et al: "Modeling positional effects of regulatory sequences with spline transformations increases prediction accuracy of deep neural networks". \'
', name=None, version='0.1', license='MIT', tags=['RNA binding'], contributors=[Author(name='Ziga Avsec', github='avsecz', email=None)], cite_as='https://doi.org/10.1093/bioinformatics/btx727', trained_on='RBP occupancy peaks measured by eCLIP-seq (Van Nostrand et al., 2016 - https://doi.org/10.1038/nmeth.3810), https://github.com/gagneurlab/Manuscript_Avsec_Bioinformatics_2017 ', training_procedure='Single task training with ADAM')
model.info.version
'0.1'
Schema
dict(model.schema.inputs)
{'dist_exon_intron': ArraySchema(shape=(1, 10), doc='Distance the nearest exon_intron (splice donor) site transformed with B-splines', name='dist_exon_intron', special_type=None, associated_metadata=[], column_labels=None),
'dist_gene_end': ArraySchema(shape=(1, 10), doc='Distance the nearest gene end transformed with B-splines', name='dist_gene_end', special_type=None, associated_metadata=[], column_labels=None),
'dist_gene_start': ArraySchema(shape=(1, 10), doc='Distance the nearest gene start transformed with B-splines', name='dist_gene_start', special_type=None, associated_metadata=[], column_labels=None),
'dist_intron_exon': ArraySchema(shape=(1, 10), doc='Distance the nearest intron_exon (splice acceptor) site transformed with B-splines', name='dist_intron_exon', special_type=None, associated_metadata=[], column_labels=None),
'dist_polya': ArraySchema(shape=(1, 10), doc='Distance the nearest Poly-A site transformed with B-splines', name='dist_polya', special_type=None, associated_metadata=[], column_labels=None),
'dist_start_codon': ArraySchema(shape=(1, 10), doc='Distance the nearest start codon transformed with B-splines', name='dist_start_codon', special_type=None, associated_metadata=[], column_labels=None),
'dist_stop_codon': ArraySchema(shape=(1, 10), doc='Distance the nearest stop codon transformed with B-splines', name='dist_stop_codon', special_type=None, associated_metadata=[], column_labels=None),
'dist_tss': ArraySchema(shape=(1, 10), doc='Distance the nearest TSS site transformed with B-splines', name='dist_tss', special_type=None, associated_metadata=[], column_labels=None),
'seq': ArraySchema(shape=(101, 4), doc='One-hot encoded RNA sequence', name='seq', special_type=<ArraySpecialType.DNASeq: 'DNASeq'>, associated_metadata=[], column_labels=None)}
model.schema.targets
ArraySchema(shape=(1,), doc='Predicted binding strength', name=None, special_type=None, associated_metadata=[], column_labels=None)
Default dataloader
Model already has the default dataloder present. To use it, specify
model.source_dir
'/home/avsec/.kipoi/models/rbp_eclip/XRCC6'
model.default_dataloader
dataloader.SeqDistDataset
model.default_dataloader.info
Info(authors=[Author(name='Ziga Avsec', github='avsecz', email=None)], doc='RBP binding model taking as input 101nt long sequence as well as 8 distances to nearest genomic landmarks - tss, poly-A, exon-intron boundary, intron-exon boundary, start codon, stop codon, gene start, gene end
', name=None, version='0.1', license='MIT', tags=[])
Predict_on_batch
model.predict_on_batch
<bound method KerasModel.predict_on_batch of <kipoi.model.KerasModel object at 0x7f95b27af2b0>>
Others
# Model source
model.source
GitLFSSource(remote_url='[email protected]:kipoi/models.git', local_path='/home/avsec/.kipoi/models/')
# model location directory
model.source_dir
'/home/avsec/.kipoi/models/rbp_eclip/XRCC6'
DataLoader
DataLoader = kipoi.get_dataloader_factory(MODEL)
# same as DataLoader = model.default_dataloader
A dataloader will most likely require input arguments in which the input files are defined, for example input fasta files or bed files, based on which the model input is generated. There are several options where the dataloader input keyword arguments are displayed:
# Display information about the dataloader
print(DataLoader.__doc__)
Args:
intervals_file: file path; tsv file
Assumes bed-like `chrom start end id score strand` format.
fasta_file: file path; Genome sequence
gtf_file: file path; Genome annotation GTF file.
filter_protein_coding: Considering genomic landmarks only for protein coding genes
preproc_transformer: file path; tranformer used for pre-processing.
target_file: file path; path to the targets
batch_size: int
# Alternatively the dataloader keyword arguments can be displayed using the function:
kipoi.print_dl_kwargs(DataLoader)
Keyword argument: `intervals_file`
doc: bed6 file with `chrom start end id score strand` columns
type: str
optional: False
example: example_files/intervals.bed
Keyword argument: `fasta_file`
doc: Reference genome sequence
type: str
optional: False
example: example_files/hg38_chr22.fa
Keyword argument: `gtf_file`
doc: file path; Genome annotation GTF file
type: str
optional: False
example: example_files/gencode.v24.annotation_chr22.gtf
Keyword argument: `filter_protein_coding`
doc: Considering genomic landmarks only for protein coding genes when computing the distances to the nearest genomic landmark.
type: str
optional: True
example: True
Keyword argument: `target_file`
doc: path to the targets (txt) file
type: str
optional: True
example: example_files/targets.tsv
Keyword argument: `use_linecache`
doc: if True, use linecache https://docs.python.org/3/library/linecache.html to access bed file rows
type: str
optional: True
--------------------------------------------------------------------------------
Example keyword arguments are: {'intervals_file': 'example_files/intervals.bed', 'fasta_file': 'example_files/hg38_chr22.fa', 'gtf_file': 'example_files/gencode.v24.annotation_chr22.gtf', 'filter_protein_coding': True, 'target_file': 'example_files/targets.tsv'}
Run dataloader on some examples
# each dataloader already provides example files which can be used to illustrate its use:
DataLoader.example_kwargs
{'fasta_file': 'example_files/hg38_chr22.fa',
'filter_protein_coding': True,
'gtf_file': 'example_files/gencode.v24.annotation_chr22.gtf',
'intervals_file': 'example_files/intervals.bed',
'target_file': 'example_files/targets.tsv'}
import os
# cd into the source directory
os.chdir(DataLoader.source_dir)
!tree
.
├── custom_keras_objects.py -> ../template/custom_keras_objects.py
├── dataloader_files
│ └── position_transformer.pkl
├── dataloader.py -> ../template/dataloader.py
├── dataloader.yaml -> ../template/dataloader.yaml
├── example_files -> ../template/example_files
├── model_files
│ └── model.h5
├── model.yaml -> ../template/model.yaml
└── __pycache__
├── custom_keras_objects.cpython-36.pyc
└── dataloader.cpython-36.pyc
4 directories, 8 files
dl = DataLoader(**DataLoader.example_kwargs)
# could be also done with DataLoader.init_example()
# This particular dataloader is of type Dataset
# i.e. it implements the __getitem__ method:
dl[0].keys()
dict_keys(['inputs', 'targets', 'metadata'])
dl[0]["inputs"]["seq"][:5]
array([[0.25, 0.25, 0.25, 0.25],
[0.25, 0.25, 0.25, 0.25],
[0.25, 0.25, 0.25, 0.25],
[0.25, 0.25, 0.25, 0.25],
[0.25, 0.25, 0.25, 0.25]], dtype=float32)
dl[0]["inputs"]["seq"][:5]
array([[0.25, 0.25, 0.25, 0.25],
[0.25, 0.25, 0.25, 0.25],
[0.25, 0.25, 0.25, 0.25],
[0.25, 0.25, 0.25, 0.25],
[0.25, 0.25, 0.25, 0.25]], dtype=float32)
len(dl)
14
Get the whole dataset
whole_data = dl.load_all()
100%|██████████| 1/1 [00:00<00:00, 6.24it/s]
whole_data.keys()
dict_keys(['inputs', 'targets', 'metadata'])
whole_data["inputs"]["seq"].shape
(14, 101, 4)
Get the iterator to run predictions
it = dl.batch_iter(batch_size=1, shuffle=False, num_workers=0, drop_last=False)
next(it)["inputs"]["seq"].shape
(1, 101, 4)
model.predict_on_batch(next(it)["inputs"])
array([[0.1351]], dtype=float32)
Pipeline
Pipeline object will take the dataloader arguments and run the whole pipeline directly:
dataloader arguments --Dataloader--> numpy arrays --Model--> prediction
example_kwargs = model.default_dataloader.example_kwargs
preds = model.pipeline.predict_example()
preds
100%|██████████| 1/1 [00:00<00:00, 6.78it/s]
array([[0.4208],
[0.0005],
[0.0005],
[0.4208],
[0.4208],
[0.4208],
[0.0005],
[0.4208],
[0.4208],
[0.4208],
[0.4208],
[0.4208],
[0.4208],
[0.4208]], dtype=float32)
model.pipeline.predict(example_kwargs)
1it [00:01, 1.56s/it]
array([0.4208, 0.0005, 0.0005, 0.4208, 0.4208, 0.4208, 0.0005, 0.4208, 0.4208, 0.4208, 0.4208,
0.4208, 0.4208, 0.4208], dtype=float32)
next(model.pipeline.predict_generator(example_kwargs, batch_size=2))
array([[0.4208],
[0.0005]], dtype=float32)
from kipoi_utils.data_utils import numpy_collate
numpy_collate_concat(list(model.pipeline.predict_generator(example_kwargs)))
array([[0.4208],
[0.0005],
[0.0005],
[0.4208],
[0.4208],
[0.4208],
[0.0005],
[0.4208],
[0.4208],
[0.4208],
[0.4208],
[0.4208],
[0.4208],
[0.4208]], dtype=float32)
Re-train the Keras model
Keras model is stored under the .model attribute.
model.model.compile("adam", "binary_crossentropy")
train_it = dl.batch_train_iter(batch_size=2)
# model.model.summary()
model.model.fit_generator(train_it, steps_per_epoch=3, epochs=1)
Epoch 1/1
3/3 [==============================] - 1s 291ms/step - loss: 1.3592
<keras.callbacks.History at 0x7f95b0095fd0>