Generated from notebooks/variant_effect_prediction.ipynb
Variant effect prediction
Variant effect prediction offers a simple way to predict effects of SNVs using any model that uses DNA sequence as an input. Many different scoring methods can be chosen, but the principle relies on in-silico mutagenesis. The default input is a VCF and the default output again is a VCF annotated with predictions of variant effects.
For details please take a look at the documentation in Postprocessing/Variant effect prediction. This iPython notebook goes through the basic programmatic steps that are needed to preform variant effect prediction. First a variant-centered approach will be taken and secondly overlap-based variant effect prediction will be presented. For details in how this is done programmatically, please refer to the documentation.
Variant centered effect prediction
Models that accept input .bed files can make use of variant-centered effect prediction. This procedure starts out from the query VCF and generates genomic regions of the length of the model input, centered on the individual variant in the VCF.The model dataloader is then used to produce the model input samples for those regions, which are then mutated according to the alleles in the VCF:
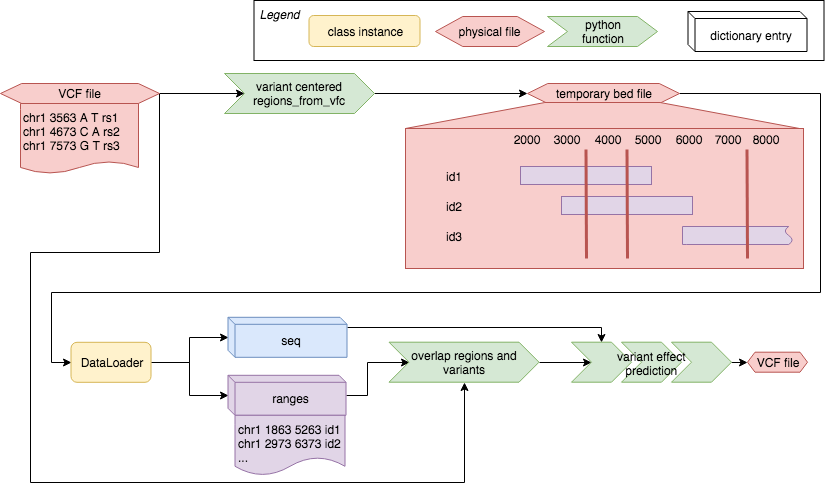
First an instance of SnvCenteredRg generates a temporary bed file with regions matching the input sequence length defined in the model.yaml input schema. Then the model dataloader is used to preduce the model input in batches. These chunks of data are then modified by the effect prediction algorithm, the model batch prediction function is triggered for all mutated sequence sets and finally the scoring method is applied.
The selected scoring methods compare model predicitons for sequences carrying the reference or alternative allele. Those scoring methods can be Diff for simple subtraction of prediction, Logit for substraction of logit-transformed model predictions, or DeepSEA_effect which is a combination of Diff and Logit, which was published in the Troyanskaya et al. (2015) publication.
This ipython notebook assumes that it is executed in an environment in which all dependencies for the following models are installed: DeepSEA/vaariantEffects, HAL, labranchor, MaxEntScan, and rbp are installed, as well as the --vep flag has to be used during installing the dependencies. Now let's start out by loading the DeepSEA model and dataloader factory:
import kipoi
model_name = "DeepSEA/variantEffects"
# get the model
model = kipoi.get_model(model_name)
# get the dataloader factory
Dataloader = kipoi.get_dataloader_factory(model_name)
Next we will have to define the variants we want to look at, let's look at a sample VCF in chromosome 22:
!head -n 40 example_data/clinvar_donor_acceptor_chr22.vcf
##fileformat=VCFv4.0
##FILTER=<ID=PASS,Description="All filters passed">
##contig=<ID=chr1,length=249250621>
##contig=<ID=chr2,length=243199373>
##contig=<ID=chr3,length=198022430>
##contig=<ID=chr4,length=191154276>
##contig=<ID=chr5,length=180915260>
##contig=<ID=chr6,length=171115067>
##contig=<ID=chr7,length=159138663>
##contig=<ID=chr8,length=146364022>
##contig=<ID=chr9,length=141213431>
##contig=<ID=chr10,length=135534747>
##contig=<ID=chr11,length=135006516>
##contig=<ID=chr12,length=133851895>
##contig=<ID=chr13,length=115169878>
##contig=<ID=chr14,length=107349540>
##contig=<ID=chr15,length=102531392>
##contig=<ID=chr16,length=90354753>
##contig=<ID=chr17,length=81195210>
##contig=<ID=chr18,length=78077248>
##contig=<ID=chr19,length=59128983>
##contig=<ID=chr20,length=63025520>
##contig=<ID=chr21,length=48129895>
##contig=<ID=chr22,length=51304566>
##contig=<ID=chrX,length=155270560>
##contig=<ID=chrY,length=59373566>
##contig=<ID=chrMT,length=16569>
#CHROM POS ID REF ALT QUAL FILTER INFO
chr22 41320486 4 G T . . .
chr22 31009031 9 T G . . .
chr22 43024150 15 C G . . .
chr22 43027392 16 A G . . .
chr22 37469571 122 C T . . .
chr22 37465112 123 C G . . .
chr22 37494466 124 G T . . .
chr22 18561373 177 G T . . .
chr22 51065593 241 C T . . .
chr22 51064006 242 C T . . .
chr22 51065269 243 G A . . .
chr22 30032866 260 G T . . .
Now we will define path variable for vcf input and output paths and instantiate a VcfWriter, which will write out the annotated VCF:
import kipoi_veff
from kipoi_veff import VcfWriter
# The input vcf path
vcf_path = "example_data/clinvar_donor_acceptor_chr22.vcf"
# The output vcf path, based on the input file name
out_vcf_fpath = vcf_path[:-4] + "%s.vcf"%model_name.replace("/", "_")
# The writer object that will output the annotated VCF
writer = VcfWriter(model, vcf_path, out_vcf_fpath)
Then we need to instantiate an object that can generate variant-centered regions (SnvCenteredRg objects). This class needs information on the model input sequence length which is extracted automatically within ModelInfoExtractor objects:
# Information extraction from dataloader and model
model_info = kipoi_veff.ModelInfoExtractor(model, Dataloader)
# vcf_to_region will generate a variant-centered regions when presented a VCF record.
vcf_to_region = kipoi_veff.SnvCenteredRg(model_info)
Now we can define the required dataloader arguments, omitting the intervals_file as this will be replaced by the automatically generated bed file:
dataloader_arguments = {"fasta_file": "example_data/hg19_chr22.fa"}
This is the moment to run the variant effect prediction:
import kipoi_veff.snv_predict as sp
from kipoi_veff.scores import Diff, DeepSEA_effect
sp.predict_snvs(model,
Dataloader,
vcf_path,
batch_size = 32,
dataloader_args=dataloader_arguments,
vcf_to_region=vcf_to_region,
evaluation_function_kwargs={'diff_types': {'diff': Diff("mean"), 'deepsea_effect': DeepSEA_effect("mean")}},
sync_pred_writer=writer)
100%|██████████| 14/14 [00:34<00:00, 2.46s/it]
In the example above we have used the variant scoring method Diff and DeepSEA_effect from kipoi_veff plug-in. As mentioned above variant scoring methods calculate the difference between predictions for reference and alternative, but there is another dimension to this: Models that have the use_rc: true flag set in their model.yaml file (DeepSEA/variantEffects does) will not only be queried with the reference and alternative carrying input sequences, but also with the reverse complement of the the sequences. In order to know of to combine predictions for forward and reverse sequences there is a initialisation flag (here set to: "mean") for the variant scoring methods. "mean" in this case means that after calculating the effect (e.g.: Difference) the average over the difference between the prediction for the forward and for the reverse sequence should be returned. Setting "mean" complies with what was used in the Troyanskaya et al. publication.
Now let's look at the output:
# Let's print out the first 40 lines of the annotated VCF (up to 80 characters per line maximum)
with open("example_data/clinvar_donor_acceptor_chr22DeepSEA_variantEffects.vcf") as ifh:
for i,l in enumerate(ifh):
long_line = ""
if len(l)>80:
long_line = "..."
print(l[:80].rstrip() +long_line)
if i >=40:
break
##fileformat=VCFv4.0
##INFO=<ID=KV:kipoi:DeepSEA/variantEffects:DIFF,Number=.,Type=String,Description...
##INFO=<ID=KV:kipoi:DeepSEA/variantEffects:DEEPSEA_EFFECT,Number=.,Type=String,D...
##INFO=<ID=KV:kipoi:DeepSEA/variantEffects:rID,Number=.,Type=String,Description=...
##FILTER=<ID=PASS,Description="All filters passed">
##contig=<ID=chr1,length=249250621>
##contig=<ID=chr2,length=243199373>
##contig=<ID=chr3,length=198022430>
##contig=<ID=chr4,length=191154276>
##contig=<ID=chr5,length=180915260>
##contig=<ID=chr6,length=171115067>
##contig=<ID=chr7,length=159138663>
##contig=<ID=chr8,length=146364022>
##contig=<ID=chr9,length=141213431>
##contig=<ID=chr10,length=135534747>
##contig=<ID=chr11,length=135006516>
##contig=<ID=chr12,length=133851895>
##contig=<ID=chr13,length=115169878>
##contig=<ID=chr14,length=107349540>
##contig=<ID=chr15,length=102531392>
##contig=<ID=chr16,length=90354753>
##contig=<ID=chr17,length=81195210>
##contig=<ID=chr18,length=78077248>
##contig=<ID=chr19,length=59128983>
##contig=<ID=chr20,length=63025520>
##contig=<ID=chr21,length=48129895>
##contig=<ID=chr22,length=51304566>
##contig=<ID=chrX,length=155270560>
##contig=<ID=chrY,length=59373566>
##contig=<ID=chrMT,length=16569>
#CHROM POS ID REF ALT QUAL FILTER INFO
chr22 41320486 4 G T . . KV:kipoi:DeepSEA/variantEffects:DIFF=-0.00285008|-0.000...
chr22 31009031 9 T G . . KV:kipoi:DeepSEA/variantEffects:DIFF=-0.02733281|-0.008...
chr22 43024150 15 C G . . KV:kipoi:DeepSEA/variantEffects:DIFF=0.01077350|0.0007...
chr22 43027392 16 A G . . KV:kipoi:DeepSEA/variantEffects:DIFF=-0.12174654|-0.24...
chr22 37469571 122 C T . . KV:kipoi:DeepSEA/variantEffects:DIFF=-0.00654625|0.00...
chr22 37465112 123 C G . . KV:kipoi:DeepSEA/variantEffects:DIFF=0.00574893|0.003...
chr22 37494466 124 G T . . KV:kipoi:DeepSEA/variantEffects:DIFF=0.01308702|0.001...
chr22 18561373 177 G T . . KV:kipoi:DeepSEA/variantEffects:DIFF=-0.00669485|0.00...
chr22 51065593 241 C T . . KV:kipoi:DeepSEA/variantEffects:DIFF=0.00409312|0.001...
chr22 51064006 242 C T . . KV:kipoi:DeepSEA/variantEffects:DIFF=0.00095593|0.000...
This shows that variants have been annotated with variant effect scores. For every different scoring method a different INFO tag was created and the score of every model output is concantenated with the | separator symbol. A legend is given in the header section of the VCF. The name tag indicates with model was used, wich version of it and it displays the scoring function label (DIFF) which is derived from the scoring function label defined in the evaluation_function_kwargs dictionary ('diff').
The most comprehensive representation of effect preditions is in the annotated VCF. Kipoi offers a VCF parser class that enables simple parsing of annotated VCFs:
from kipoi_veff.parsers import KipoiVCFParser
vcf_reader = KipoiVCFParser("example_data/clinvar_donor_acceptor_chr22DeepSEA_variantEffects.vcf")
#We can have a look at the different labels which were created in the VCF
print(list(vcf_reader.kipoi_parsed_colnames.values()))
[('kipoi', 'DeepSEA/variantEffects', 'DIFF'), ('kipoi', 'DeepSEA/variantEffects', 'DEEPSEA_EFFECT'), ('kipoi', 'DeepSEA/variantEffects', 'rID')]
We can see that two scores have been saved - 'DEEPSEA_EFFECT' and 'DIFF'. Additionally there is 'rID' which is the region ID - that is the ID given by the dataloader for a genomic region which was overlapped with the variant to get the prediction that is listed in the effect score columns mentioned before. Let's take a look at the VCF entries:
import pandas as pd
entries = [el for el in vcf_reader]
print(pd.DataFrame(entries).head().iloc[:,:7])
variant_id variant_chr variant_pos variant_ref variant_alt \
0 4 chr22 41320486 G T
1 9 chr22 31009031 T G
2 15 chr22 43024150 C G
3 16 chr22 43027392 A G
4 122 chr22 37469571 C T
KV_DeepSEA/variantEffects_DIFF_8988T_DNase_None_0 \
0 -0.002850
1 -0.027333
2 0.010774
3 -0.121747
4 -0.006546
KV_DeepSEA/variantEffects_DIFF_AoSMC_DNase_None_1
0 -0.000094
1 -0.008740
2 0.000702
3 -0.247321
4 0.000784
Another way to access effect predicitons programmatically is to keep all the results in memory and receive them as a dictionary of pandas dataframes:
effects = sp.predict_snvs(model,
Dataloader,
vcf_path,
batch_size = 32,
dataloader_args=dataloader_arguments,
vcf_to_region=vcf_to_region,
evaluation_function_kwargs={'diff_types': {'diff': Diff("mean"), 'deepsea_effect': DeepSEA_effect("mean")}},
return_predictions=True)
100%|██████████| 14/14 [00:33<00:00, 2.41s/it]
For every key in the evaluation_function_kwargs dictionary there is a key in effects and (the equivalent of an additional INFO tag in the VCF). Now let's take a look at the results:
for k in effects:
print(k)
print(effects[k].head().iloc[:,:4])
print("-"*80)
diff
8988T_DNase_None AoSMC_DNase_None \
chr22:41320486:G:['T'] -0.002850 -0.000094
chr22:31009031:T:['G'] -0.027333 -0.008740
chr22:43024150:C:['G'] 0.010773 0.000702
chr22:43027392:A:['G'] -0.121747 -0.247321
chr22:37469571:C:['T'] -0.006546 0.000784
Chorion_DNase_None CLL_DNase_None
chr22:41320486:G:['T'] -0.001533 -0.000353
chr22:31009031:T:['G'] -0.003499 -0.008143
chr22:43024150:C:['G'] 0.004689 -0.000609
chr22:43027392:A:['G'] -0.167689 -0.010695
chr22:37469571:C:['T'] -0.000383 -0.000924
--------------------------------------------------------------------------------
deepsea_effect
8988T_DNase_None AoSMC_DNase_None \
chr22:41320486:G:['T'] 0.000377 9.663903e-07
chr22:31009031:T:['G'] 0.004129 3.683221e-03
chr22:43024150:C:['G'] 0.001582 1.824510e-04
chr22:43027392:A:['G'] 0.068382 2.689577e-01
chr22:37469571:C:['T'] 0.001174 4.173280e-04
Chorion_DNase_None CLL_DNase_None
chr22:41320486:G:['T'] 0.000162 0.000040
chr22:31009031:T:['G'] 0.000201 0.002139
chr22:43024150:C:['G'] 0.000322 0.000033
chr22:43027392:A:['G'] 0.133855 0.000773
chr22:37469571:C:['T'] 0.000008 0.000079
--------------------------------------------------------------------------------
We see that for diff and deepsea_effect there is a dataframe with variant identifiers as rows and model output labels as columns. The DeepSEA model predicts 919 tasks simultaneously hence there are 919 columns in the dataframe.
Overlap based prediction
Models that cannot predict on every region of the genome might not accept a .bed file as dataloader input. An example of such a model is a splicing model. Those models only work in certain regions of the genome. Here variant effect prediction can be executed based on overlaps between the regions generated by the dataloader and the variants defined in the VCF:
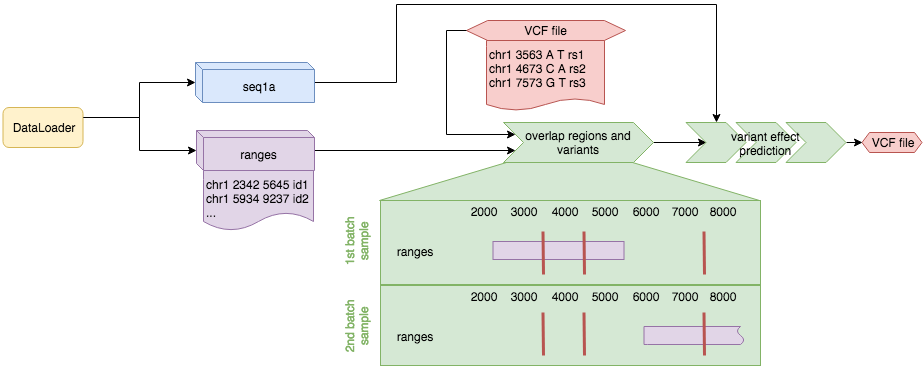 The procedure is similar to the variant centered effect prediction explained above, but in this case no temporary bed file is generated and the effect prediction is based on all the regions generated by the dataloader which overlap any variant in the VCF. If a region is overlapped by two variants the effect of the two variants is predicted independently.
The procedure is similar to the variant centered effect prediction explained above, but in this case no temporary bed file is generated and the effect prediction is based on all the regions generated by the dataloader which overlap any variant in the VCF. If a region is overlapped by two variants the effect of the two variants is predicted independently.
Here the VCF has to be tabixed so that a regional lookup can be performed efficiently, this can be done by using the ensure_tabixed function, the rest remains the same as before:
import kipoi
from kipoi_veff import VcfWriter
from kipoi_veff import ensure_tabixed_vcf
# Use a splicing model
model_name = "HAL"
# get the model
model = kipoi.get_model(model_name)
# get the dataloader factory
Dataloader = kipoi.get_dataloader_factory(model_name)
# The input vcf path
vcf_path = "example_data/clinvar_donor_acceptor_chr22.vcf"
# Make sure that the vcf is bgzipped and tabixed, if not then generate the compressed vcf in the same place
vcf_path_tbx = ensure_tabixed_vcf(vcf_path)
# The output vcf path, based on the input file name
out_vcf_fpath = vcf_path[:-4] + "%s.vcf"%model_name.replace("/", "_")
# The writer object that will output the annotated VCF
writer = VcfWriter(model, vcf_path, out_vcf_fpath)
This time we don't need an object that generates regions, hence we can directly define the dataloader arguments and run the prediction:
from kipoi_veff import predict_snvs
from kipoi_veff.scores import Diff
dataloader_arguments = {"gtf_file":"example_data/Homo_sapiens.GRCh37.75.filtered_chr22.gtf",
"fasta_file": "example_data/hg19_chr22.fa"}
effects = predict_snvs(model,
Dataloader,
vcf_path_tbx,
batch_size = 32,
dataloader_args=dataloader_arguments,
evaluation_function_kwargs={'diff_types': {'diff': Diff("mean")}},
sync_pred_writer=writer,
return_predictions=True)
100%|██████████| 709/709 [00:04<00:00, 150.84it/s]
Let's have a look at the VCF:
# A slightly convoluted way of printing out the first 40 lines and up to 80 characters per line maximum
with open("example_data/clinvar_donor_acceptor_chr22HAL.vcf") as ifh:
for i,l in enumerate(ifh):
long_line = ""
if len(l)>80:
long_line = "..."
print(l[:80].rstrip() +long_line)
if i >=40:
break
##fileformat=VCFv4.0
##INFO=<ID=KV:kipoi:HAL:DIFF,Number=.,Type=String,Description="DIFF SNV effect p...
##INFO=<ID=KV:kipoi:HAL:rID,Number=.,Type=String,Description="Range or region id...
##FILTER=<ID=PASS,Description="All filters passed">
##contig=<ID=chr1,length=249250621>
##contig=<ID=chr2,length=243199373>
##contig=<ID=chr3,length=198022430>
##contig=<ID=chr4,length=191154276>
##contig=<ID=chr5,length=180915260>
##contig=<ID=chr6,length=171115067>
##contig=<ID=chr7,length=159138663>
##contig=<ID=chr8,length=146364022>
##contig=<ID=chr9,length=141213431>
##contig=<ID=chr10,length=135534747>
##contig=<ID=chr11,length=135006516>
##contig=<ID=chr12,length=133851895>
##contig=<ID=chr13,length=115169878>
##contig=<ID=chr14,length=107349540>
##contig=<ID=chr15,length=102531392>
##contig=<ID=chr16,length=90354753>
##contig=<ID=chr17,length=81195210>
##contig=<ID=chr18,length=78077248>
##contig=<ID=chr19,length=59128983>
##contig=<ID=chr20,length=63025520>
##contig=<ID=chr21,length=48129895>
##contig=<ID=chr22,length=51304566>
##contig=<ID=chrX,length=155270560>
##contig=<ID=chrY,length=59373566>
##contig=<ID=chrMT,length=16569>
#CHROM POS ID REF ALT QUAL FILTER INFO
chr22 17684454 4461 G A . . KV:kipoi:HAL:DIFF=0.10586491;KV:kipoi:HAL:rID=290
chr22 17669232 7178 T C . . KV:kipoi:HAL:DIFF=0.00000000;KV:kipoi:HAL:rID=293
chr22 17669232 7178 T C . . KV:kipoi:HAL:DIFF=0.00000000;KV:kipoi:HAL:rID=299
chr22 17684454 4461 G A . . KV:kipoi:HAL:DIFF=0.10586491;KV:kipoi:HAL:rID=304
chr22 17669232 7178 T C . . KV:kipoi:HAL:DIFF=0.00000000;KV:kipoi:HAL:rID=307
chr22 17684454 4461 G A . . KV:kipoi:HAL:DIFF=0.10586491;KV:kipoi:HAL:rID=313
chr22 17669232 7178 T C . . KV:kipoi:HAL:DIFF=0.00000000;KV:kipoi:HAL:rID=316
chr22 17684454 4461 G A . . KV:kipoi:HAL:DIFF=0.10586491;KV:kipoi:HAL:rID=322
chr22 17669232 7178 T C . . KV:kipoi:HAL:DIFF=0.00000000;KV:kipoi:HAL:rID=325
chr22 17669232 7178 T C . . KV:kipoi:HAL:DIFF=0.00000000;KV:kipoi:HAL:rID=328
chr22 18561370 7302 C T . . KV:kipoi:HAL:DIFF=-1.33794269;KV:kipoi:HAL:rID=824
And the prediction output this time is less helpful because it's the ids that the dataloader created which are displayed as index. In general it is advisable to use the output VCF for more detailed information on which variant was overlapped with which region fo produce a prediction.
for k in effects:
print(k)
print(effects[k].head())
print("-"*80)
diff
0
290 0.105865
293 0.000000
299 0.000000
304 0.105865
307 0.000000
--------------------------------------------------------------------------------
Command-line based effect prediction
The above command can also conveniently be executed using the command line:
import json
import os
model_name = "DeepSEA/variantEffects"
dl_args = json.dumps({"fasta_file": "example_data/hg19_chr22.fa"})
out_vcf_fpath = vcf_path[:-4] + "%s.vcf"%model_name.replace("/", "_")
scorings = "diff deepsea_effect"
command = ("kipoi veff score_variants {model} "
"--dataloader_args='{dl_args}' "
"-i {input_vcf} "
"-o {output_vcf} "
"-s {scorings}").format(model=model_name,
dl_args=dl_args,
input_vcf=vcf_path,
output_vcf=out_vcf_fpath,
scorings=scorings)
# Print out the command:
print(command)
kipoi veff score_variants DeepSEA/variantEffects --dataloader_args='{"fasta_file": "example_data/hg19_chr22.fa"}' -i example_data/clinvar_donor_acceptor_chr22.vcf -o example_data/clinvar_donor_acceptor_chr22DeepSEA_variantEffects.vcf -s diff deepsea_effect
! $command
[32mINFO[0m [44m[kipoi.sources][0m Update /nfs/research1/stegle/users/rkreuzhu/.kipoi/models/[0m
Already up-to-date.
[32mINFO[0m [44m[kipoi.sources][0m git-lfs pull -I DeepSEA/variantEffects/**[0m
[32mINFO[0m [44m[kipoi.sources][0m git-lfs pull -I DeepSEA/template/**[0m
[32mINFO[0m [44m[kipoi.sources][0m git-lfs pull -I DeepSEA/template/model_files/**[0m
[32mINFO[0m [44m[kipoi.sources][0m git-lfs pull -I DeepSEA/template/example_files/**[0m
[32mINFO[0m [44m[kipoi.sources][0m model DeepSEA/variantEffects loaded[0m
[32mINFO[0m [44m[kipoi.sources][0m git-lfs pull -I DeepSEA/variantEffects/./**[0m
[32mINFO[0m [44m[kipoi.sources][0m git-lfs pull -I DeepSEA/template/**[0m
[32mINFO[0m [44m[kipoi.sources][0m git-lfs pull -I DeepSEA/template/model_files/**[0m
[32mINFO[0m [44m[kipoi.sources][0m git-lfs pull -I DeepSEA/template/example_files/**[0m
[32mINFO[0m [44m[kipoi.sources][0m dataloader DeepSEA/variantEffects/. loaded[0m
[32mINFO[0m [44m[kipoi.data][0m successfully loaded the dataloader from /nfs/research1/stegle/users/rkreuzhu/.kipoi/models/DeepSEA/variantEffects/dataloader.py::SeqDataset[0m
[32mINFO[0m [44m[kipoi.pipeline][0m dataloader.output_schema is compatible with model.schema[0m
100%|███████████████████████████████████████████| 14/14 [00:35<00:00, 2.53s/it]
Batch prediction
Since the syntax basically doesn't change for different kinds of models a simple for-loop can be written to do what we just did on many models:
import kipoi
# Run effect predicton
models_df = kipoi.list_models()
models_substr = ["HAL", "MaxEntScan", "labranchor", "rbp"]
models_df_subsets = {ms: models_df.loc[models_df["model"].str.contains(ms)] for ms in models_substr}
/nfs/research1/stegle/users/rkreuzhu/opt/model-zoo/kipoi/config.py:110: FutureWarning: Sorting because non-concatenation axis is not aligned. A future version
of pandas will change to not sort by default.
To accept the future behavior, pass 'sort=False'.
To retain the current behavior and silence the warning, pass 'sort=True'.
return pd.concat(pd_list)[pd_list[0].columns]
# Run variant effect prediction using a basic Diff
import kipoi
from kipoi_veff import ensure_tabixed_vcf
import kipoi_veff.snv_predict as sp
from kipoi_veff import VcfWriter
from kipoi_veff.scores import Diff
splicing_dl_args = {"gtf_file":"example_data/Homo_sapiens.GRCh37.75.filtered_chr22.gtf",
"fasta_file": "example_data/hg19_chr22.fa"}
dataloader_args_dict = {"HAL": splicing_dl_args,
"labranchor": splicing_dl_args,
"MaxEntScan":splicing_dl_args,
"rbp": {"fasta_file": "example_data/hg19_chr22.fa",
"gtf_file":"example_data/Homo_sapiens.GRCh37.75_chr22.gtf"}
}
for ms in models_substr:
model_name = models_df_subsets[ms]["model"].iloc[0]
#kipoi.pipeline.install_model_requirements(model_name)
model = kipoi.get_model(model_name)
vcf_path = "example_data/clinvar_donor_acceptor_chr22.vcf"
vcf_path_tbx = ensure_tabixed_vcf(vcf_path)
out_vcf_fpath = vcf_path[:-4] + "%s.vcf"%model_name.replace("/", "_")
writer = VcfWriter(model, vcf_path, out_vcf_fpath)
print(model_name)
Dataloader = kipoi.get_dataloader_factory(model_name)
dataloader_arguments = dataloader_args_dict[ms]
model_info = kipoi_veff.ModelInfoExtractor(model, Dataloader)
vcf_to_region = None
if ms == "rbp":
vcf_to_region = kipoi_veff.SnvCenteredRg(model_info)
sp.predict_snvs(model,
Dataloader,
vcf_path_tbx,
batch_size = 32,
dataloader_args=dataloader_arguments,
vcf_to_region=vcf_to_region,
evaluation_function_kwargs={'diff_types': {'diff': Diff("mean")}},
sync_pred_writer=writer)
writer.close()
HAL
100%|██████████| 709/709 [00:04<00:00, 167.58it/s]
MaxEntScan/3prime
100%|██████████| 709/709 [00:02<00:00, 329.18it/s]
Using TensorFlow backend.
WARNING:tensorflow:From /nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/keras/backend/tensorflow_backend.py:1238: calling reduce_sum (from tensorflow.python.ops.math_ops) with keep_dims is deprecated and will be removed in a future version.
Instructions for updating:
keep_dims is deprecated, use keepdims instead
WARNING:tensorflow:From /nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/keras/backend/tensorflow_backend.py:1340: calling reduce_mean (from tensorflow.python.ops.math_ops) with keep_dims is deprecated and will be removed in a future version.
Instructions for updating:
keep_dims is deprecated, use keepdims instead
/nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/keras/models.py:287: UserWarning: Error in loading the saved optimizer state. As a result, your model is starting with a freshly initialized optimizer.
warnings.warn('Error in loading the saved optimizer '
labranchor
100%|██████████| 709/709 [00:05<00:00, 122.95it/s]
2018-07-25 11:37:28,705 [INFO] successfully loaded the dataloader from /nfs/research1/stegle/users/rkreuzhu/.kipoi/models/rbp_eclip/AARS/dataloader.py::SeqDistDataset
WARNING:tensorflow:From /nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/keras/backend/tensorflow_backend.py:1255: calling reduce_prod (from tensorflow.python.ops.math_ops) with keep_dims is deprecated and will be removed in a future version.
Instructions for updating:
keep_dims is deprecated, use keepdims instead
2018-07-25 11:37:28,851 [WARNING] From /nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/keras/backend/tensorflow_backend.py:1255: calling reduce_prod (from tensorflow.python.ops.math_ops) with keep_dims is deprecated and will be removed in a future version.
Instructions for updating:
keep_dims is deprecated, use keepdims instead
/nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/keras/models.py:287: UserWarning: Error in loading the saved optimizer state. As a result, your model is starting with a freshly initialized optimizer.
warnings.warn('Error in loading the saved optimizer '
2018-07-25 11:37:30,996 [INFO] successfully loaded the model from model_files/model.h5
2018-07-25 11:37:30,999 [INFO] dataloader.output_schema is compatible with model.schema
2018-07-25 11:37:31,157 [INFO] git-lfs pull -I rbp_eclip/AARS/**
rbp_eclip/AARS
2018-07-25 11:37:32,071 [INFO] git-lfs pull -I rbp_eclip/template/**
2018-07-25 11:37:33,219 [INFO] dataloader rbp_eclip/AARS loaded
2018-07-25 11:37:33,252 [INFO] successfully loaded the dataloader from /nfs/research1/stegle/users/rkreuzhu/.kipoi/models/rbp_eclip/AARS/dataloader.py::SeqDistDataset
2018-07-25 11:37:34,411 [INFO] Extracted GTF attributes: ['gene_id', 'gene_name', 'gene_source', 'gene_biotype', 'transcript_id', 'transcript_name', 'transcript_source', 'exon_number', 'exon_id', 'tag', 'protein_id', 'ccds_id']
/nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/sklearn/base.py:311: UserWarning: Trying to unpickle estimator MinMaxScaler from version 0.18.1 when using version 0.19.1. This might lead to breaking code or invalid results. Use at your own risk.
UserWarning)
/nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/sklearn/base.py:311: UserWarning: Trying to unpickle estimator Imputer from version 0.18.1 when using version 0.19.1. This might lead to breaking code or invalid results. Use at your own risk.
UserWarning)
0%| | 0/14 [00:00<?, ?it/s]INFO:2018-07-25 11:37:34,812:genomelake] Running landmark extractors..
2018-07-25 11:37:34,812 [INFO] Running landmark extractors..
/nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/concise/utils/position.py:55: FutureWarning: from_items is deprecated. Please use DataFrame.from_dict(dict(items), ...) instead. DataFrame.from_dict(OrderedDict(items)) may be used to preserve the key order.
("strand", gtf.strand)])
/nfs/research1/stegle/users/rkreuzhu/conda-envs/kipoi_interpret/lib/python3.6/site-packages/concise/utils/position.py:62: FutureWarning: from_items is deprecated. Please use DataFrame.from_dict(dict(items), ...) instead. DataFrame.from_dict(OrderedDict(items)) may be used to preserve the key order.
("strand", gtf.strand)])
INFO:2018-07-25 11:37:34,975:genomelake] Done!
2018-07-25 11:37:34,975 [INFO] Done!
100%|██████████| 14/14 [00:03<00:00, 4.05it/s]
let's validate that things have worked:
! wc -l example_data/clinvar_donor_acceptor_chr22*.vcf
450 example_data/clinvar_donor_acceptor_chr22DeepSEA_variantEffects.vcf
2035 example_data/clinvar_donor_acceptor_chr22HAL.vcf
794 example_data/clinvar_donor_acceptor_chr22labranchor.vcf
1176 example_data/clinvar_donor_acceptor_chr22MaxEntScan_3prime.vcf
449 example_data/clinvar_donor_acceptor_chr22rbp_eclip_AARS.vcf
447 example_data/clinvar_donor_acceptor_chr22.vcf
5351 total