| Model name | Version | Authors | Contributed by | Type | Cite as | License | Training procedure |
|---|---|---|---|---|---|---|---|
| Model name | Version | Authors | Contributed by | Type | Cite as | License | Training procedure |
| MMSplice/deltaLogitPSI | 0.1 | Jun Cheng | Jun Cheng | custom | MIT | ||
| MMSplice/modularPredictions | 0.1 | Jun Cheng | Jun Cheng | custom | MIT | ||
| MMSplice/mtsplice | 0.1 | Jun Cheng | Jun Cheng | custom | MIT | ||
| MMSplice/pathogenicity | 0.1 | Jun Cheng | Jun Cheng | custom | MIT | ||
| MMSplice/splicingEfficiency | 0.1 | Jun Cheng | Jun Cheng | custom | MIT |
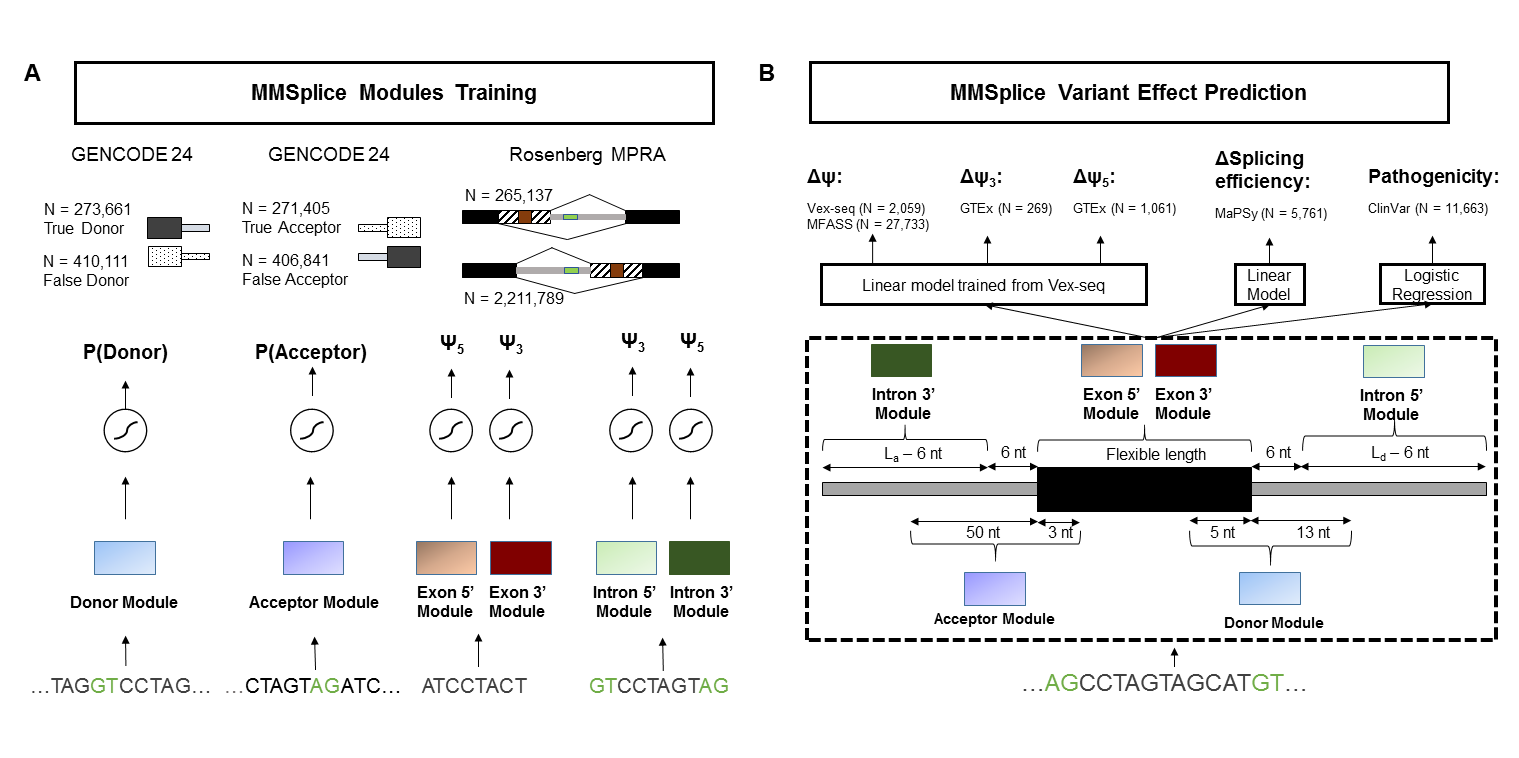
Modular Modeling of Splicing (MMSplice)
MMSplice predicts variant effect with 5 modules scoring exon, donor, acceptor, 3' intron and 5' intron. Modular predictions are combined with a linear model to predict $\Delta logit(\Psi)$ or with a logistic regression model to predict variant pathogenicity.
To score variants (including indels), we suggest to use primarily the deltaLogitPSI predictions.
Quick start example code
Install kipoi as described in: http://kipoi.org/docs/#installation
- Create a new conda environment with all dependencies installed
kipoi env create MMSplice/deltaLogitPSI source activate kipoi-MMSplice__deltaLogitPSI -
Install model dependencies into current environment
kipoi env install MMSplice/deltaLogitPSI -
Run deltaLogitPSI prediction on example files from the command line with
kipoi predict. MMSplice takes three input files: a gtf annotation file, a reference sequence fasta file and a vcf file with variants.
cd ~/.kipoi/models/MMSplice/deltaLogitPSI
kipoi predict MMSplice/deltaLogitPSI \
--dataloader_args='{'gtf_file': 'example_files/test.gtf', 'fasta_file': 'example_files/hg19.nochr.chr17.fa', 'vcf_file': 'example_files/test.vcf.gz'}' \ -o test.tsv
# check the results
head 'test.tsv'
We also documented the usage with CLI, python and R here.
Prepare your own input files
1. Prepare annotation (gtf) file
Standard human gene annotation file in GTF format can be downloaded from ensembl or gencode.
MMSplice can work directly with those files, however, some filtering is higly recommended.
- Filter for protein coding genes.
- Filter out duplicated exons. The same exon can be annotated multiple times if it appears in multiple transcripts. This will cause duplicated predictions.
We provide a filtered version here.
Note this version has chromosome names in the format chr*. You may need to remove them to match the chromosome names in your fasta file.
2. Prepare variant (VCF) file
A correctly formatted VCF file will work with MMSplice, however the following steps will make it less prone to false positives:
- Quality filtering. Low quality variants lead to unreliable predictions.
- Avoid presenting multiple variants in one line by splitting them into multiple lines. Example code to do this:
bcftools norm -m-both -o out.vcf in.vcf.gz - Left-normalization. For instance, GGCA-->GG is not left-normalized while GCA-->G is. Details on unified representation of genetic variants see Tan et al.
bcftools norm -f reference.fasta -o out.vcf in.vcf
3. Prepare reference genome (fasta) file
Human reference fasta file can be downloaded from ensembl/gencode. Make sure the chromosome names match with the GTF annotation file you use.
Repository
This repository hosts following models:
deltaLogitPSI: predict $\Delta logit(\Psi)$. Returns one prediction per variant-exon pair.
pathogenicity: predict variant pathogenicity. Returns one prediction per variant.
splicingEfficiency: predict splicing efficiency changes. Returns one prediction per variant-exon pair. Splicing efficiency model were trained from MMSplice modules and exonic variants from MaPSy, thus only the predictions for exonic variants are calibrated.
modularPredictions: the raw predictions from the five modules for reference sequence and alternative sequence. Returns a vector of length 10 for each variant-exon pair.
modules: contains individual modules for exon, donor, acceptor, 3' intron and 5' intron.
deltaLogitPSI, splicingEfficiency and pathogenicity differ by the last modular combination model.
Publication
Paper: Cheng et al. https://doi.org/10.1101/438986
Package: https://github.com/gagneurlab/MMSplice